IRB Manager
-
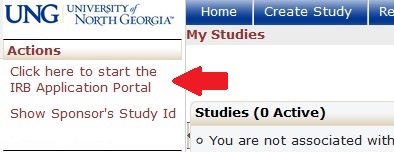
After logging into IRB Manager (UNG IRB Manager Login), click the link in the left hand column entitled “Click here to start the IRB Application Portal.” From there you will have the option to start a new regular IRB application (Form 1.1), a new alternative application (Form 1.5), or a notification about course-related research (Form 2.1). The IRB Application Portal also includes a tool to determine which application is required for your projects, as well as tool to determine which CITI Training modules are required for your research.
-
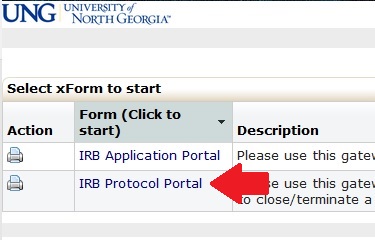
For existing studies approved by the IRB, you must navigate to that study’s page on IRB Manager to submit forms for that study. A listing of studies you are associated with are on your IRB Manager homepage/dashboard under the “My Studies” section. To navigate to the study’s page, click on the study number. When you are on the study’s page, click the “Start xForm” link in the left had column, and click the link entitled “IRB Protocol Portal.” Once you are in the IRB Protocol Portal, you have the option to begin forms for your study, including forms for continuing review (Form 1.2), notification of completion of a study (Form 1.3), research modifications (Form 1.4), and reporting of an unanticipated problem or adverse event (Form 4.1).
-
Yes. Each form has a header section, which includes a drop down menu to the various pages of each form. Using this drop down menu, you can view the main questions on each page. Please note that answers to these main questions may trigger additional questions asked.

-
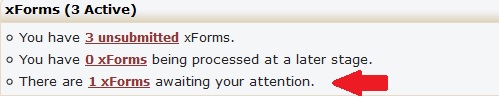
Once you can begin a form, your progress will automatically be saved, and you can return to the form and continue to edit any response to a question before submitting the form. You can find your forms in progress on your IRB Manager homepage/dashboard under the “xForms” section. Those forms will be marked “unsubmitted.”
-
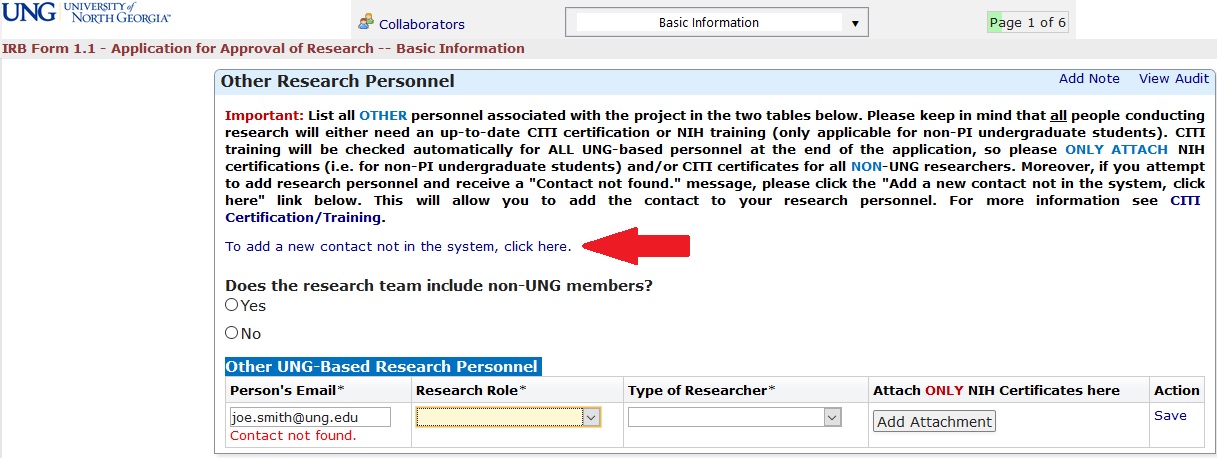
If you receive a “Contact not found” error message, you can add this contact by clicking the link entitled “To add a new contact not in the system, click here.” This will open up the Add Contact form which will enable you to add the contact’s information into IRB Manager. Once you have submitted the Add Contact form, you can return to your application and add the contact to the Research Personnel table.
-
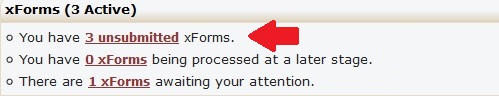
In the email you receive requesting the changes or clarifications to your application, there will be a link to your application. Clicking that link will allow you make any changes necessary, including attaching any additional or revised documents. You can also find any form that has been returned for changes or clarifications on your IRB Manager home page/dashboard. Those forms will be in the “xForms” section and will be mark as “awaiting your attention.” Please ensure you click the “Submit” button on the last page once you have completed your changes. Failure to click the “Submit” button will result in the IRB not receiving your changes and could delay your application’s review.